Role of 16S rRNA and Next Generation Sequencing (NGS) in Classifying a Microbial Species
Hello Steemians,
Discovery of the Gene Sequencing plays a major role in classifying any new bacterial species and adding it to the literature. As in my last post, some of the readers could not get a full sight of it so I thought to explain it properly. In today’s article I will be covering what are the housekeeping genes, what importance they possess, which are considered to be the housekeeping genes in bacteria, how to isolate these gene and further sequence it through the genome sequencer.

What are Housekeeping Genes?
Housekeeping gene are named so as they are present in all cells of all tissue irrespective of the specificity of the tissue and play a major role in regulating all the metabolic process from DNA replication to cellular byproducts. These genes are also called as constitutive genes: genes which are essential for the functioning of cell and expressing all the time. (1,2) Microbial species have several constitutive genes some of them are 16S Ribosomal RNA, DNA Gyrase A, Recombinase A, DNA-directed RNA polymerase (α and β subunits), Glyceraldehyde-3-phosphate dehydrogenase, protein translocase and many more. Among them 16S rRNA is most widely used for the identification purpose as these genes have a slow rate of evolution over time, other genes are also used for differentiating between species but not that frequent. As the 16S rRNA gene is coding for the 30S subunit of the ribosome that specifically binds on the Shine-Dalgarno sequence.(3)
16S rRNA Gene (The Molecular Clock)
It’s almost 1500 base pair four domain RNA sequence act as a platform for the biding of ribosomal protein. Its 3’-end bind to the start codon of mRNA and initiates the protein synthesis. 16S rRNA also known to act as mediator for the binding of both the ribosomal subunits and regulates the correct pairing of codon-anticodons. The 1500 bp sequence has about 14 universal primers and among them 27F (forward) and 1492R (reverse) are used for the first Polymerase Chain Reaction (PCR) amplification, which amplifies this specific region from the whole genome of the bacteria. These 16S rRNA genes contains hypervariable region which can be even used to differentiate between two very closely related species (4). This advancement of 16S rRNA gene gave a cheap and very efficient alternative for the identification of bacteria.
Isolating the 16S rRNA gene
A microbial species which has to be identified is grown freshly and the genomic DNA of it get extracted by any of the efficient method. Then the extracted genomic DNA first checked for the purity by either running it on agarose gel or through NanoDrop. Agarose gel will show the intense band in the region of the genomic DNA size of that bacterial species or closely related species and through NanoDrop absorbance at 260/280nm show the contamination of protein in the extracted sample. After verifying the extracted genomic DNA, it can be used for the PCR amplification. As the whole region is of around 1500 basepairs, two primers of the extreme ends can be used to amplify the whole region. For those who don’t know much about PCR (Polymerase Chain Reaction), it’s an amplification process which includes 3 major steps of;
- Denaturation
- Annaeling
- Elongation
In the first step the double stranded DNA got denatured at high temperature 94°C and the two single strands are than get annealed with the two forward and reverse primers. The temperature for this step varies according to the G+C content (Guanosine:Cytosine) of the DNA molecule. In the last step of elongation, the two annealed primers starts adding up the nucleotides onto the single strands at 72°C with the help of polymerase enzyme (Taq polymerase). This whole process is of about 2-3 minutes depending on the annealing time and about 30 cycles of it can provide you a million copy of the amplified region.
 Standard 16S rRNA PCR Amplification
Standard 16S rRNA PCR AmplificationDNA Sequencing (Whole Genome Sequencing)
DNA sequencing deals with a process which involves the determination of nucleotide position and location in the genome. DNA sequence is made from four different nucleotides Adenine, Guanine, Cytosine and Thymine and their order of arrangement possess the key to synthesize the correct amino acid and which further chained up to form a functional protein. DNA sequence has a wide range of application starting from the molecular biology of genes which encodes the sequence for a protein. Metagenomics which deals with the identification of a large population from a specific source which gives idea of a group of population present in that environment. Further applications in Medicine and Forensics which may help to cure several diseases and also help in the forensic identification. DNA sequencing can also be used to sequence the whole genome of an organism.
Whole genome sequencing is the process which involves the sequencing of an organism genome. The genome analysis has attracted almost doctors and researchers from all the branch, as it give all the information of the organism. Gene encode the proteins and proteins plays all the role in an organism body, whole genome has all the necessary information one wants. Whole genome also helps in determining the evolutionary lineage of an organism by the phylogenic study. It started in early 1970s and 1980s two manual method process were given Maxam-Gilbert Sequencing and Sanger Sequencing. Till date Sanger sequencing principle is being followed up and being an efficient one, with this a project to sequence the human genome has also started named as Human Genome Project (HGP), it was launched in 1990 and last up to 2003. In 2004 they published an incomplete version of this project.
Next Generation Sequencing (NGS)
Apart from the Maxam-Gilbert and Sanger sequencing several new method were developed for synthesizing the nucleotide in the mid-1990s and were commercialized by 2000. These new techniques for sequencing the genome called as Next Genome Sequencing. There are several basic methods involved in the sequencing like Maxam-Gilbert which involves the radioactive labelling of DNA molecule and is bit complex, Chain termination method also called as Sanger’s method for sequencing was a manual method but after its great success it became automated and during this period several advancement has made in this field of genome sequencing. 454 pyrosequencing, Polony sequencing, illumina (Solexa) sequencing, Ion torrent semiconductor identification, single molecule real time (SMRT) sequencing and Nanopore DNA sequencing are some of the examples of the new techniques involved in next generation sequencing. But all these deals with the sequencing of the genomic DNA which makes different contigs of the genome and sequence them individually. The sequenced contigs can further be arranged among themselves and whole genome is designed.

Wikimedia Commons Illumina HiSeq 2500
But for determining the sequence of a small region like 16S rRNA there is no need to identify the whole genome of the microbe. The amplified region of 16S rRNA act as a contig from the genomic DNA and their primers may give you the sequence of the gene through chain termination process. For determining the bacteria species by sequencing 16S rRNA region different primers can be used. The amplified product from the first PCR process mixed with the sequencing buffer and the TRR (Termination Reaction Ready: A mixture containing enzyme and the ddNTPs). ddNTPs (dideoxy Nucleotide Triphosphate) is known to terminate the reaction, it can sense the region where the nucleotides is having end codons. The result of the sequenced gene comes out like (in image) and these individual color peaks corresponds to the different nucleotide.
Methodology of the Sequencer
It’s something like, a single strand DNA is present and when a new nucleotide gets added up into the system it gives a color signal which produces a colored chromatogram. The whole process works like the gene you provide to the instrument uses a primer and the primer sits on one of the end based on the forward or reverse sequence and then from a bottle which contains all the four nucleotide passes the all four nucleotide one by one and the one which is complementary to the sequence will bind to the template and emits color which was recorded as the attachment of nucleotide. The intensity of the chromatogram peak show the purity of the DNA sequence and hence gives a confirmed sequence. Two peaks in a single slot shows binding of two nucleotide to the single complementary nucleotide shows the contaminated DNA or mixture of two genes together. Most of the time not maintaining the molar ratio can result in the no binding of nucleotide and shows blank with no peaks.
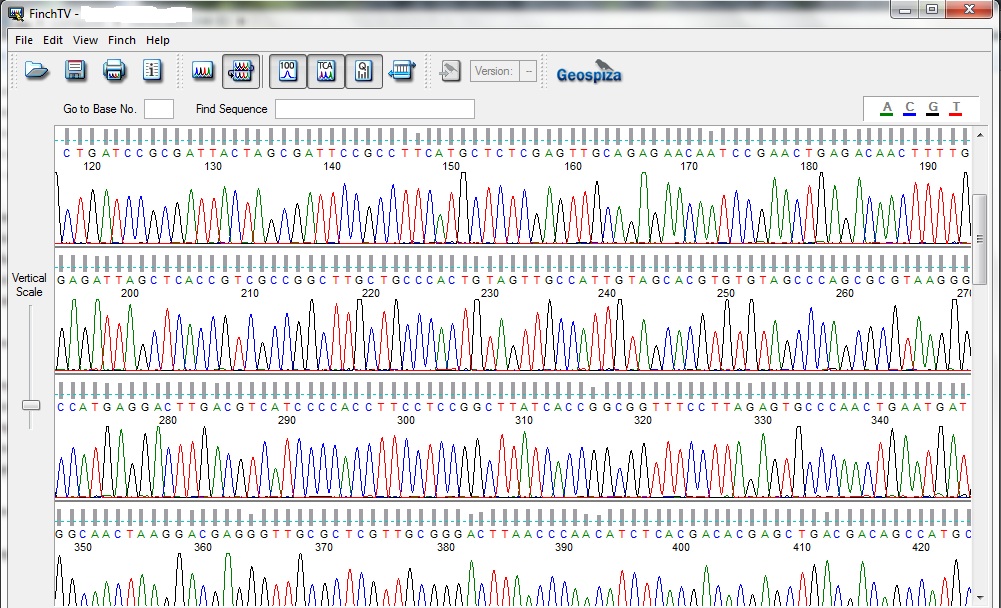
The sequenced gene (16S rRNA) was further processed to determine the similarity and identify the differences it has, for that we need to select the sequence from the chromatogram file and as I mentioned if you see and mixture of peaks and any sort of irregularity in the peak, it will always result in the improper result. Better to use a sequence which shows a clean and sharp chromatogram. The selected gene from the sequence file was further uploaded on the servers which aligned the sequence with all the reported genome and according to the similarity index it will show you the best match. NCBI BLAST (Basic Local Alignment Search Tool) and EzCloud can be used to do the above task. The same sequence can be used to make the phylogenetic tree from the acquired DNA sequence.
Determining the Novel species from this Data
16S rRNA is the best way till date to identify the novel microorganism. The percentage identity match less than 97% could be a potential novel species and conserved gene sequence can also be used to give a novel genus by differentiating characteristics from some related species, just on the basis of 16S rRNA also new species can be proposed.
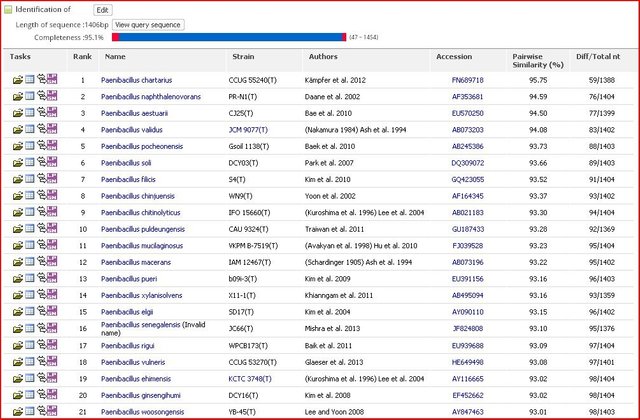
Alignment of the sequenced DNA with the reported Type strain sequences through EzTaxon. Pairwise similarity of 95% showing a potential novel species.
The variation occur very frequently in between 30-100 base pair and change in only few nucleotide position could sometime result in the production of organism from two different niche. Sometime metagenomic study can also give you some novel species as a part of isolating the same group of bacteria.
For most of the terms I have hyperlinked with the simple Wikipedia page, that will make you understand if not feel free to ask anything below in the comment section. I will be happy to have them and will try to resolve doubts if anyone have.
Images which are not sourced are taken by me and do not violates any copyright.
References
Eisenberg et. al., 2013 Housekeeping genes, revisited. (https://www.ncbi.nlm.nih.gov/pubmed/23810203)
Goldman et. al., 2001 Housekeeping Genes. (https://www.sciencedirect.com/science/article/pii/B012227080000639X)
Danilo et. al., 2015 Bacterial reference genes for gene expression studies by RT-qPCR: survey and analysis. (https://link.springer.com/article/10.1007%2Fs10482-015-0524-1)
Wikipedia : 16S rRNA (https://en.wikipedia.org/wiki/16S_ribosomal_RNA)
Wikipedia : Polymerase Chain Reaction (https://en.wikipedia.org/wiki/Polymerase_chain_reaction)
Wikipedia : DNA Sequencing (https://en.wikipedia.org/wiki/DNA_sequencing)
Hi @micro24
We have selected your post as post of the day for our DaVinci Times. Our goal is to help the scientific community of Steemit, and even if our vote is still small we hope to grow in quickly! You will soon receive our sincere upvote! If you are interested in science follow us sto learn more about our project.
Immagine CC0 Creative Commons, si ringrazia @mrazura per il logo ITASTEM.
CLICK HERE AND VOTE FOR DAVINCI.WITNESS
Keep in mind that for organizational reasons it’s necessary to use the “steemstem” and “davinci-times” tags to be voted again.
Greetings from @davinci.witness and the itaSTEM team.
Nice one @micro24
As far as I can remember, this Paenibacillus sp. was really a potential one, with the novelty it also shows the AMP activity. So, what's the status of this?
Actually bacteriocines they produces are sort of narrow range and are effective on the gram negatives only and the whole genome also didn't gave much information. Its now in the middle and we are doing some bioinformatic work on its genome to find something as it is showing about 95% similarity.
Thanks, this gives a very clear answer to the question I asked in your other article.
You are most welcome @flyyingkiwi